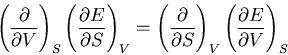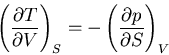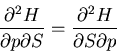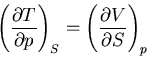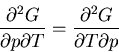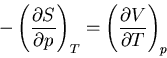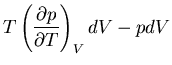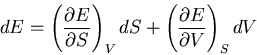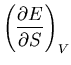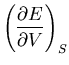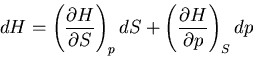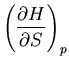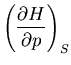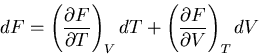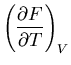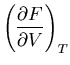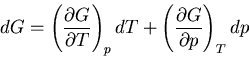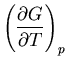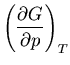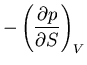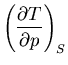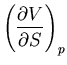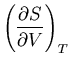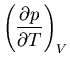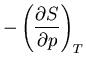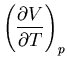Next: About this document ...
LECTURE 7
General Relations for a Homogeneous Substance
For simplicity we will omit the averaging bar; macroscopic quantities
such as energy 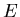 and pressure
and pressure 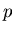 will be understood to refer to their
mean values. A macrostate can be specified by 2 macroscopic variables.
For example if we specify the volume
will be understood to refer to their
mean values. A macrostate can be specified by 2 macroscopic variables.
For example if we specify the volume 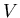 and internal energy , then
the other macroscopic parameters such as and
and internal energy , then
the other macroscopic parameters such as and 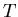 are then determined.
We usually like to specify the macroscopic parameters which we can
control in the lab or in our simulation. So and may not be
the most convenient. We can pick any 2 macroscopic parameters, like
and , and then determine the others (e.g., and
are then determined.
We usually like to specify the macroscopic parameters which we can
control in the lab or in our simulation. So and may not be
the most convenient. We can pick any 2 macroscopic parameters, like
and , and then determine the others (e.g., and 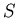 ). To determine
the other macroscopic parameters, we need to know their relation to
the ones that we specified. Let us now derive these relations.
In doing so, we will define a number of useful quantities such as
enthalpy, the Helmholtz free energy and the Gibbs free energy.
). To determine
the other macroscopic parameters, we need to know their relation to
the ones that we specified. Let us now derive these relations.
In doing so, we will define a number of useful quantities such as
enthalpy, the Helmholtz free energy and the Gibbs free energy.
We start with the fundamental thermodynamic relation for a quasi-static
infinitesimal process:
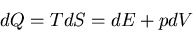 |
(1) |
Summary of Maxwell relations and thermodynamic functions
These are Maxwell's relations which we derived starting from the
fundamental relation
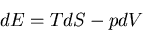 |
(42) |
Notice that we can rewrite this
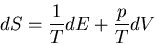 |
(43) |
Since the entropy is a state function, 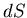 is an exact differential
and we can write it as:
is an exact differential
and we can write it as:
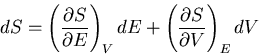 |
(44) |
Comparing coefficients of 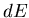 and
and 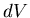 , we find
, we find
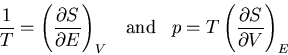 |
(45) |
These are relations that we derived earlier. Recall
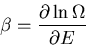 |
(46) |
and
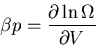 |
(47) |
Note that the conjugate pairs of variables
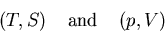 |
(48) |
appear paired in (42) and when one cross multiplies the
Maxwell relations. In other words the numerator on one side is conjugate to
the denominator on the other side. To obtain the correct sign, note that
if the two variables with respect to which one differentiates are the same
variables and which occur as differentials in (42),
then the minus sign that occurs in (42) also occurs in the
Maxwell relation. Any one permutation away from these particular variables
introduces a change of sign. For example, consider the Maxwell relation
(39)
with derivatives with respect to and . Switching from to
implies one sign change with respect to the minus sign in (42);
hence there is a plus sign in (39).
We also summarize the thermodynamic functions:
Notice that the conjugate variables are always paired up. The sign changes
when one changes the independent variable compared to the fundamental
relation (42).
Phase Transitions and the Clausius-Clapeyron Equation
Let me try to give some idea of why these thermodynamic functions
are useful. We know from classical mechanics and electromagnetism
that energy is a very useful concept because it is conserved, and
because systems try to minimize their energy. Now we have ``generalized
energies'' such as 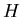 ,
, 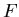 , and
, and 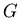 . Chemists like enthalpy
because in the lab pressure rather than volume is kept constant.
Note that
. Chemists like enthalpy
because in the lab pressure rather than volume is kept constant.
Note that 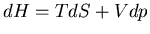 , so if pressure is constant which means
, so if pressure is constant which means
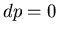 , then
, then 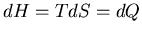 which is why enthalpy is thought of as heat.
When physicists refer to free energy, they usually mean the Helmholtz
free energy
which is why enthalpy is thought of as heat.
When physicists refer to free energy, they usually mean the Helmholtz
free energy 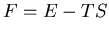 because they can calculate this starting
from the Hamiltonian. However, experimentally it is the Gibbs free
energy
because they can calculate this starting
from the Hamiltonian. However, experimentally it is the Gibbs free
energy 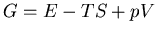 that is relevant. Physicists often speak about
minimizing the
free energy. Consider water and ice. If we were to just consider
minimizing the internal energy which is the sum of the kinetic
and potential energies, then ice would be the lowest energy state.
Water molecules have less kinetic energy in ice than in water. Also
the water molecules are, on average, farther from each other in ice than
in water, so their potential energy of interaction is less in ice than
in water. But ice only exists at low temperatures. Why is that?
Because at high temperatures the free energy of water is lower than
that of ice. If we consider constant pressure and hence the Gibbs
free energy, then we want to minimize
that is relevant. Physicists often speak about
minimizing the
free energy. Consider water and ice. If we were to just consider
minimizing the internal energy which is the sum of the kinetic
and potential energies, then ice would be the lowest energy state.
Water molecules have less kinetic energy in ice than in water. Also
the water molecules are, on average, farther from each other in ice than
in water, so their potential energy of interaction is less in ice than
in water. But ice only exists at low temperatures. Why is that?
Because at high temperatures the free energy of water is lower than
that of ice. If we consider constant pressure and hence the Gibbs
free energy, then we want to minimize
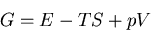 |
(57) |
At high temperatures the second term 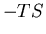 is important. Water molecules
have a much higher entropy in their liquid state than in their solid
state. More entropy means more microstates in phase space which improves
the chances of the system being in a liquid microstate. (Just like buying
more lottery tickets improves your chances of winning.)
The higher entropy of the liquid offsets the fact that
is important. Water molecules
have a much higher entropy in their liquid state than in their solid
state. More entropy means more microstates in phase space which improves
the chances of the system being in a liquid microstate. (Just like buying
more lottery tickets improves your chances of winning.)
The higher entropy of the liquid offsets the fact that
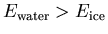 . At low
temperatures is less important, so
matters and the free energy for ice is lower than that of water. The
transition temperature
. At low
temperatures is less important, so
matters and the free energy for ice is lower than that of water. The
transition temperature 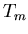 between ice and water is given by
between ice and water is given by
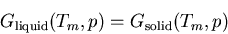 |
(58) |
We can actually use this relation to derive an interesting relation
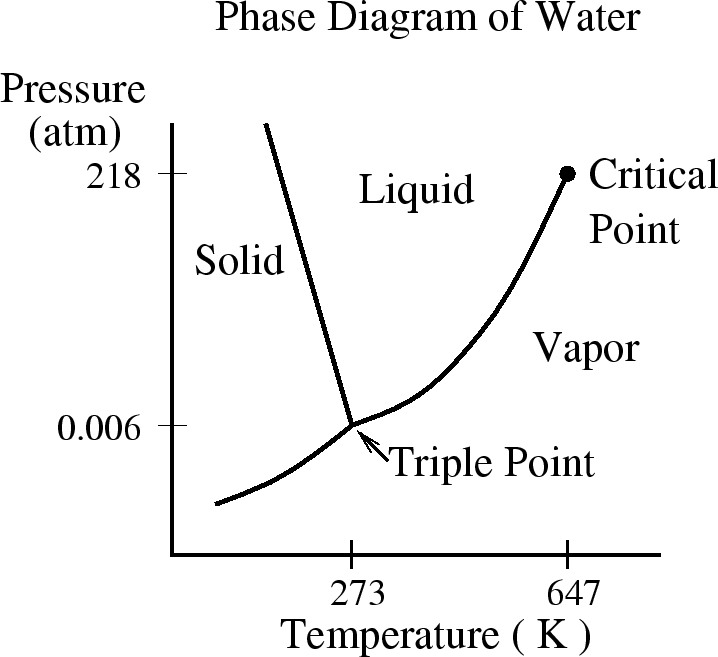
called the Clausius-Clapeyron equation. Remember I told you that
water is unusual because ice expands and because the slope of the
phase boundary between ice and water is negative ( ). These
facts are related through the Clausius-Claperyon equation. (See Reif 8.5
for more details.)
). These
facts are related through the Clausius-Claperyon equation. (See Reif 8.5
for more details.)
=3.0 true in
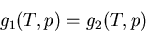 |
(59) |
Here
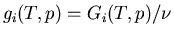 is the Gibbs free energy per mole of phase
is the Gibbs free energy per mole of phase
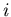 at temperature and pressure . If we move a little ways along
the phase boundary, then we have
at temperature and pressure . If we move a little ways along
the phase boundary, then we have
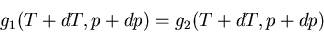 |
(60) |
Subtracting these two equations leads to the condition:
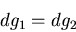 |
(61) |
Now use (57)
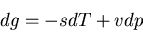 |
(62) |
where 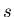 is the molar entropy and
is the molar entropy and 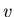 is the molar volume.
So (62) becomes
is the molar volume.
So (62) becomes
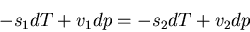 |
(63) |
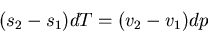 |
(64) |
or
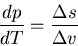 |
(65) |
where
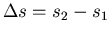 and
and
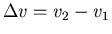 . This is called the
Clausius-Clapeyron equation. It relates the slope of the phase
boundary at a given point to the ratio of the entropy change
. This is called the
Clausius-Clapeyron equation. It relates the slope of the phase
boundary at a given point to the ratio of the entropy change
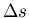 to the volume change
to the volume change 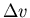 .
.
Let's apply this to
the water-ice transition. We know that the slope of the phase
boundary . Let phase 1 be water and let phase 2 be ice.
Then
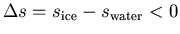 since ice has less entropy
than water. Putting these 2 facts together in
the Clausius-Clapeyron equation implies that we must have
since ice has less entropy
than water. Putting these 2 facts together in
the Clausius-Clapeyron equation implies that we must have 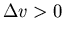 .
Indeed water expands on freezing and
.
Indeed water expands on freezing and
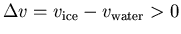 .
So the unusual negative slope of the melting line means that water
expands on freezing. As we mentioned earlier, it also means that you
can cool down a water-ice mixture by pressurizing it and following
the coexistence curve, i.e., the melting line or phase boundary.
.
So the unusual negative slope of the melting line means that water
expands on freezing. As we mentioned earlier, it also means that you
can cool down a water-ice mixture by pressurizing it and following
the coexistence curve, i.e., the melting line or phase boundary.
Another example is 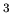 He which also has a melting line with a negative
slope. The fact that means that if you increase the pressure
on a mixture of liquid and solid He, the temperature will drop. This
is the principle behind cooling with a Pomeranchuk cell. Unlike the case
of water where ice floats because it is less dense, solid He sinks
because it is more dense than liquid He. The Clausius-Clapeyron
equation then implies that
He which also has a melting line with a negative
slope. The fact that means that if you increase the pressure
on a mixture of liquid and solid He, the temperature will drop. This
is the principle behind cooling with a Pomeranchuk cell. Unlike the case
of water where ice floats because it is less dense, solid He sinks
because it is more dense than liquid He. The Clausius-Clapeyron
equation then implies that
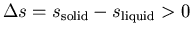 ,
i.e., solid He has more entropy than liquid He! How can this be?
It turns out that it is spin entropy. A He atom is a fermion with a nuclear
spin 1/2. The atoms in liquid He roam around and their wavefunctions
overlap, so that liquid He has a Fermi sea just like electrons do.
The Fermi energy is lower if the He atoms can pair up with opposite spins
so that two He atoms can occupy each translational energy state.
However, in solid liquid He, the atoms are centered on lattice
sites and the wavefunctions do not overlap much. So the spins on
different atoms in the solid are not correlated and the spin
entropy is
,
i.e., solid He has more entropy than liquid He! How can this be?
It turns out that it is spin entropy. A He atom is a fermion with a nuclear
spin 1/2. The atoms in liquid He roam around and their wavefunctions
overlap, so that liquid He has a Fermi sea just like electrons do.
The Fermi energy is lower if the He atoms can pair up with opposite spins
so that two He atoms can occupy each translational energy state.
However, in solid liquid He, the atoms are centered on lattice
sites and the wavefunctions do not overlap much. So the spins on
different atoms in the solid are not correlated and the spin
entropy is 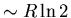 which is much larger than in the liquid.
which is much larger than in the liquid.
Now back to the general case. Since there is an entropy change associated with
the phase transformation from phase 1 to phase 2, heat must be absorbed
(or emitted). The ``latent heat of transformation'' 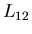 is defined
as the heat absorbed when a given amount of phase 1 is transformed to
phase 2. For example, to melt a solid, you dump heat into it
until it
reaches the melting temperature. When it reaches the melting temperature,
its temperature stays at even though you continue to dump in heat.
It uses the absorbed heat to transform the solid into liquid. This heat is
the latent heat .
Since the process takes place at the constant temperature , the
corresponding entropy change is simply
is defined
as the heat absorbed when a given amount of phase 1 is transformed to
phase 2. For example, to melt a solid, you dump heat into it
until it
reaches the melting temperature. When it reaches the melting temperature,
its temperature stays at even though you continue to dump in heat.
It uses the absorbed heat to transform the solid into liquid. This heat is
the latent heat .
Since the process takes place at the constant temperature , the
corresponding entropy change is simply
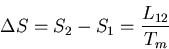 |
(66) |
Thus the Clausius-Clapeyron equation (66) can be written
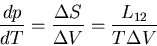 |
(67) |
If refers to the molar volume, then is the latent
heat per mole; if refers to the volume per gram, then
is the latent heat per gram. In most substances, the latent heat
is used to melt the solid into a liquid. However, if you put
heat into liquid He when it is on the melting line, it will
form solid because solid He has more entropy than liquid He.
Examples of using the Maxwell relations
Let's give some examples where the Maxwell relations can be used.
Suppose we want to calculate
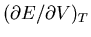 . We start
with our usual fundamental relation
. We start
with our usual fundamental relation
|
(68) |
We want to replace with . So we regard the entropy as a function
of the independent variables and :
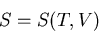 |
(69) |
is an exact differential:
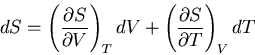 |
(70) |
Since we are interested in
at constant
temperature, 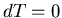 and
and
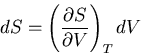 |
(71) |
Now use the Maxwell relation (40):
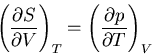 |
(72) |
So we have
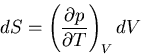 |
(73) |
and
Hence
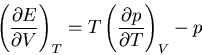 |
(75) |
Let's take a moment to consider what this means physically. We know
that gas cools when it expands, and that the pressure rises when it
is heated. There must be some connection between these two
phenomena. Microscopically we can think of the kinetic energy of the
gas molecules. Macroscopically, eq. (76) gives the relation.
If we hold the volume fixed and increase the temperature, the pressure
rises at a rate
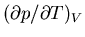 . Related to that fact is this:
if we increase the volume, the gas will cool unless we pour some heat
in to maintain the temperature, and
. Related to that fact is this:
if we increase the volume, the gas will cool unless we pour some heat
in to maintain the temperature, and
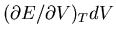 tells us the amount of heat needed to maintain the temperature.
(Notice that for an ideal gas
tells us the amount of heat needed to maintain the temperature.
(Notice that for an ideal gas
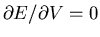 .)
Equation (76) expresses
the fundamental relation between these two effects. Notice that we
didn't need to know the microscopic interactions between the gas
particles in order to deduce the relationship between the amount of heat
needed to maintain a constant temperature when the gas expands,
and the pressure change when the gas is heated. That's what thermodynamics
does; it gives us relationships between macroscopic quantities without
having to know about microscopics.
.)
Equation (76) expresses
the fundamental relation between these two effects. Notice that we
didn't need to know the microscopic interactions between the gas
particles in order to deduce the relationship between the amount of heat
needed to maintain a constant temperature when the gas expands,
and the pressure change when the gas is heated. That's what thermodynamics
does; it gives us relationships between macroscopic quantities without
having to know about microscopics.
Aside: Notice that
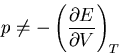 |
(76) |
as one might expect. Rather
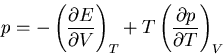 |
(77) |
However,
|
(78) |
implies that
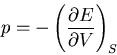 |
(79) |
It matters what is kept constant! Recall from (25) that
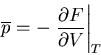 |
(80) |
Specific Heats
Consider a homogeneous substance whose volume is the only relevant
external parameter. We want the relation between the molar specific heat
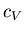 at constant volume and the molar specific heat
at constant volume and the molar specific heat 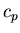 at constant
pressure. We found this relation earlier for an ideal gas
(
at constant
pressure. We found this relation earlier for an ideal gas
(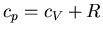 ), but now we want the general relation. This is a useful
relation because theoretical calculations are usually done at constant
volume and experimental measurements are done at constant pressure.
This is also a nice illustration of the usefulness of the Maxwell relations
and other identities and definitions.
), but now we want the general relation. This is a useful
relation because theoretical calculations are usually done at constant
volume and experimental measurements are done at constant pressure.
This is also a nice illustration of the usefulness of the Maxwell relations
and other identities and definitions.
The heat capacity at constant volume is given by
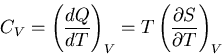 |
(81) |
and the heat capacity at constant pressure is
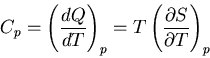 |
(82) |
Let us consider the independent variables as and . Then 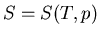 and
and
![\begin{displaymath}
dQ=TdS=T\left[\left(\frac{\partial S}{\partial T}\right)_pdT+
\left(\frac{\partial S}{\partial p}\right)_Tdp\right]
\end{displaymath}](img147.png) |
(83) |
Using (83), we have
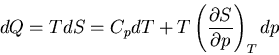 |
(84) |
At constant pressure, and we obtain (83). But to calculate
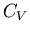 , we see from (82) that and are the independent
variables. So we plug
, we see from (82) that and are the independent
variables. So we plug
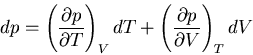 |
(85) |
into (85) to obtain
![\begin{displaymath}
dQ=TdS=C_pdT+T\left(\frac{\partial S}{\partial p}\right)_T\l...
...t)_VdT+\left(\frac{\partial p}
{\partial V}\right)_T dV\right]
\end{displaymath}](img151.png) |
(86) |
Constant means that 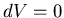 and so
and so
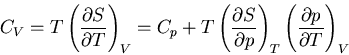 |
(87) |
This is a relation between and 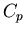 but it involves derivatives
which are not easily measured. However we can use Maxwell's relations to
write this relation in terms of quantities that are measurable.
In particular (41) is
but it involves derivatives
which are not easily measured. However we can use Maxwell's relations to
write this relation in terms of quantities that are measurable.
In particular (41) is
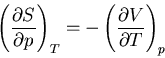 |
(88) |
The change of volume with temperature at constant pressure is related to
the ``volume coefficient of expansion'' 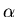 (sometimes called the
coefficient of thermal expansion):
(sometimes called the
coefficient of thermal expansion):
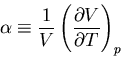 |
(89) |
Thus
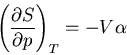 |
(90) |
The derivative
is also not easy to measure
since measurements at constant volume are difficult. It is easier to
control and . So let's write
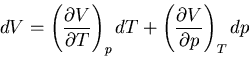 |
(91) |
For constant volume and
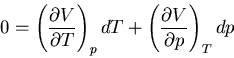 |
(92) |
we can rearrange this get an expression for 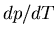 :
:
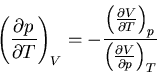 |
(93) |
Aside:
This is an example of the general relation proved in Appendix 9. If we have
3 variables 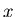 ,
, 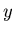 , and
, and 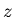 , two of which are independent, then we can
write, for example,
, two of which are independent, then we can
write, for example,
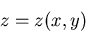 |
(94) |
and
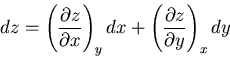 |
(95) |
At constant we have 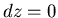 and
and
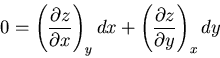 |
(96) |
Thus
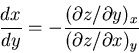 |
(97) |
or, since was kept constant
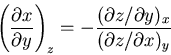 |
(98) |
This sort of relation between partial derivatives
is used extensively in thermodynamics.
Returning to (94), we note that the numerator is related to
the expansion coefficient . The denominator measures the change
in the volume of the substance with increasing pressure at constant
temperature. The change of the volume will be negative, since the volume
decreases with increasing pressure. We can define the ``isothermal
compressibility'' of the substance:
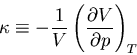 |
(99) |
The compressibility is a measure of how squishy the substance is.
Hence (94) becomes
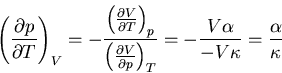 |
(100) |
Plugging (91) and (101) into (88) yields
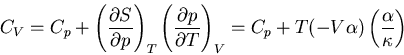 |
(101) |
or
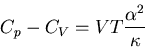 |
(102) |
Let's test this formula on the simple case of an ideal gas. We start with
the equation of state:
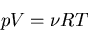 |
(103) |
We need to calculate the expansion coefficient. For constant
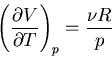 |
(104) |
Hence
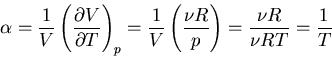 |
(105) |
Next we calculate the compressibility 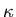 . At constant temperature
the equation of state yields
. At constant temperature
the equation of state yields
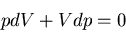 |
(106) |
Hence
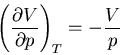 |
(107) |
and
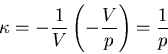 |
(108) |
Thus (103) becomes
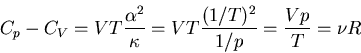 |
(109) |
or, per mole,
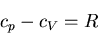 |
(110) |
which agrees with our previous result.
Limiting properties of the specific heat as
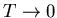
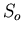 , independent of all parameters of the system. This is just
a statement that the number of states at low temperatures is very small.
In the case of a nondegenerate ground state, there is just one state and
, independent of all parameters of the system. This is just
a statement that the number of states at low temperatures is very small.
In the case of a nondegenerate ground state, there is just one state and
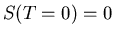 . So, in general,
. So, in general,
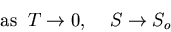 |
(111) |
This implies that the derivative
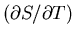 remains finite
as
. In other words, it does not go to infinity. (Technically
speaking, the derivatives appearing in (82) and (83)
remain finite as goes to 0.) So one can conclude that the
heat capacity goes to 0 as
:
remains finite
as
. In other words, it does not go to infinity. (Technically
speaking, the derivatives appearing in (82) and (83)
remain finite as goes to 0.) So one can conclude that the
heat capacity goes to 0 as
:
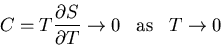 |
(112) |
or more precisely,
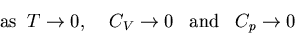 |
(113) |
The fact that the heat capacity goes to 0 at zero temperature merely
reflects the fact that the system settles into its ground state as
. If we recall that
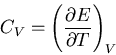 |
(114) |
then reducing the temperature further will not change the energy since the
energy has bottomed out.
Notice that we need
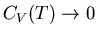 as
in order
to guarantee proper convergence of the integral in
as
in order
to guarantee proper convergence of the integral in
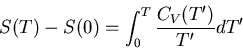 |
(115) |
The entropy difference on the left must be finite, so the integral
must also be finite.
Next: About this document ...
Clare Yu
2007-04-25