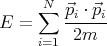
|
| (1) |
The classical partition function is
![1 ∫ { [ 1 ( ) ]}
Z ′ =--3N- exp - β ---- ⃗p21 + ⃗p22+ ...+ ⃗p2N d ⃗p1...d ⃗pNd⃗q1...d⃗qN
h o 2m](lecture111x.png) | (2) |
The reason for the prime is that this partition function is slightly defective as we shall see. The
integrals over the 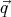 i’s gives a factor of V N. The integrals over momenta factorize into N
separate integrals. So we can write:
i’s gives a factor of V N. The integrals over momenta factorize into N
separate integrals. So we can write:
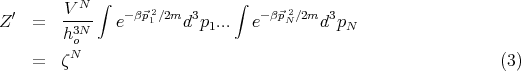
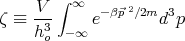 | (4) |
is the partition function for a single molecule. So
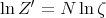 | (5) |
It is straightforward to evaluate ζ:
![∫
-V- ∞ -(β∕2m )⃗p2 3
ζ = h3 -∞ e d p
o∫ ∫ ∞ ∫
= -V- e-(β∕2m)(p2x+p2y+p2z)dpxdpydpz
h3o -∞
V [∫ ∞ 2 ]3
= --3 e-βp∕2mdp (6)
h o -∞](lecture116x.png)
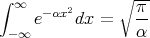 | (7) |
So we evaluate the integral:
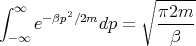 | (8) |
Hence
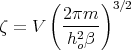 | (9) |
So
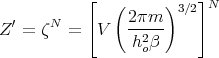 | (10) |
and
![[ ( ) ]
′ 3 3 2πm
ln Z = N ln V - --lnβ + --ln --2--
2 2 ho](lecture1111x.png) | (11) |
From our previous equations we can calculate the mean pressure p, the mean energy E, and the specific heat CV .
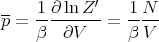 | (12) |
or
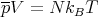 | (13) |
This is the equation of state of the ideal gas. The mean energy is given by
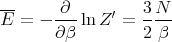 | (14) |
or
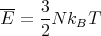 | (15) |
This is what we expect from the equipartition theorem. The heat capacity is given by
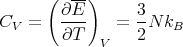 | (16) |
Using N = νNa, where Na is Avogadro’s number and ν is the number of moles, and R = NakB, we get for the molar specific heat
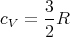 | (17) |
This agrees with our previous result.
We can also calculate the entropy S:
![[ ( ) ]
′ -- 3 3 2πm 3
S = kB [ln Z + βE ] = N kB ln V - -lnβ + --ln --2-- + --
2 2 ho 2](lecture1118x.png) | (18) |
or
![[ 3 ]
S = N kB lnV + 2-ln T + σ](lecture1119x.png) | (19) |
where
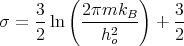 | (20) |
is a constant independent of T, V , and N. This expression for the entropy is wrong!
But there’s another problem with S. Namely the entropy S does not scale with the size of the system as an extensive parameter should. In other words if V and N double, then the S and E should double. Consider a box with an internal partition. The total entropy is S1 + S2. We know this to be true since Z = Z1Z2 if systems 1 and 2 are completely independent, as they are in this example. Thus
![--
S1 = kB [ln Z1 + βE1 ]
--
S2 = kB [ln Z2 + βE2 ] (21)](lecture1121x.png)
![S = k [lnZ Z + β(E- + E- )]
B 1 2 -- 1 2 --
= kB[lnZ1 + β E1] + kB[lnZ2 + β E2 ]
= S1 + S2 (22)](lecture1122x.png)
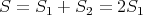 | (23) |
if the two sides are identical.
Now go back to our expression for the entropy of an ideal gas and note that
![[ ] [ ]
S (2N, 2V ) - 2S (N, V ) = 2N k ln(2V ) + 3ln T + σ - 2N k lnV + 3-lnT + σ
B 2 B 2
= 2N kB ln2
⁄= 0 (24)](lecture1124x.png)
If the gases on the two sides were different, then an increase in S when the partition function is removed seems reasonable since the two gases will diffuse and intermingle. In other words we expect the configurational entropy to increase. Then putting back the partition clearly would not restore the total system to its original state. On the other hand, if the gases are identical, then the restoration of the partition returns the system to a state physically indistinguishable from the initial state.
So if the gases on the two sides are identical, this increase in S does not make physical sense. The difficulty is associated with the concept of indistinguishability of the particles. If we exchange the positions of two identical particles, then we really haven’t changed any physical properties of the system. Yet we have counted these two identical microstates as truly distinct in calculating the entropy. (Recall S = kB ln Ω.)
This concept of indistinguishability is clearly well established in quantum mechanics where this concept is basic to the theory. We have taken the classical picture of atoms too seriously and have finally exceeded the limits of classical validity.
Gibbs postulated that the classical partition function should be divided by N! to correct for counting each indistinguishable microstate. Let
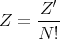 | (25) |
Then
 | (26) |
where Stirling’s approximation has been used. The entropy becomes
![--
S = kB [ln Z + βE ]
3
= kBN [ln V + 2-lnT + σ] + kB [N - N ln N ]
= kBN [ln V--+ 3-ln T + σ + 1 ] (27)
N 2](lecture1127x.png)
![V-- 3-
S = kBN [ln N + 2 ln T + σo ]](lecture1128x.png) | (28) |
where
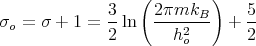 | (29) |
Now when the system is doubled (N → 2N, V → 2V ), the entropy is doubled. Later when we repeat the calculation of the entropy of an ideal monatomic gas using quantum statistics, the factor of N! will arise naturally. Also the constant ho will be identified as Planck’s constant.
It is interesting to note that this basic concept of quantum mechanics (indistinguishability of identical particles) did not arise spontaneously with the development of quantum mechanics, but was suggested by the ad hoc resolution of a paradox in classical statistical mechanics. Thus the Gibbs’ paradox joins black body radiation, the photoelectric effect, atomic line spectra and all the other pieces of evidence that led theorists to advance beyond classical mechanics to quantum mechanics.
We have used a classical approximation to describe an ideal monatomic gas. Under what conditions would we expect it to be valid? One approach to answer this question begins with the uncertainty relation:
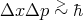 | (30) |
We expect Δx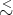 R where R is the mean separation of the ideal gas atoms, and Δp
R where R is the mean separation of the ideal gas atoms, and Δp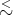 p where p
is the mean momentum. Then we would expect that classical mechanics would adequately
describe the dynamics of the gas whenever
p where p
is the mean momentum. Then we would expect that classical mechanics would adequately
describe the dynamics of the gas whenever
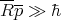 | (31) |
This effectively says that ℏ is negligible. Recall that
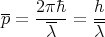 | (32) |
where λ is the mean de Broglie wavelength. Then
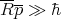 | (33) |
becomes
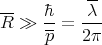 | (34) |
Or more simply, the classical description applies when
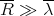 | (35) |
i.e., the mean separation between particles is much greater than their mean de Broglie wavelength.
To estimate R, imagine each molecule at the center of a little cube of side R, these cubes filling the available volume V . Then
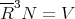 | (36) |
or
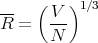 | (37) |
The mean momentum p can be estimated using the equipartition theorem
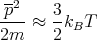 | (38) |
or
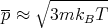 | (39) |
and
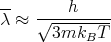 | (40) |
Hence the condition that the classical description should be valid is
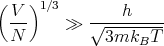 | (41) |
So for large masses and high temperatures we expect classical mechanics to hold.
As an example, air at standard temperature and pressure (STP) has T = 300 K and mean pressure P = 760 mm of Hg (mercury) = 1.01 × 105 Newton/m2. 80% of air is N 2. So the mass of a nitrogen molecule is m = 28mp where mp = 1.67 × 10-27 kilograms is the mass of a proton. Using kB = 1.38 × 10-23 Joules/K, we obtain
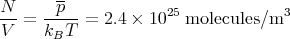 | (42) |
or
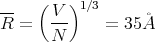 | (43) |
and
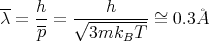 | (44) |
Thus
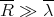 | (45) |
So the classical description holds for air. However, this is not the case for an electron gas
in a metal at 300 K. In that case we find R 2 or 3 Å and λ
2 or 3 Å and λ 60 Å (using the
electron mass). Thus λ ≫R and we must use a full quantum treatment for an electron
gas.
60 Å (using the
electron mass). Thus λ ≫R and we must use a full quantum treatment for an electron
gas.
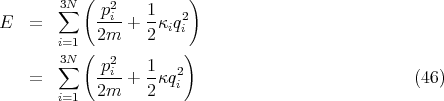

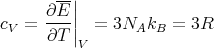 | (48) |
where NA is Avogadro’s number (NA = 6.02 × 1023 molecules/mole) or
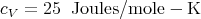 | (49) |
Notice that the specific heat is a constant independent of T. This is is called the Dulong–Petit value of the specific heat. The values of cp for some common substances at room temperature in units of Joules mole-1 K-1 are (from Reif p. 254):
| solid | cp | solid | cp |
| Cu | 24.5 | Al | 24.4 |
| Ag | 25.5 | Sn | 26.4 |
| Pb | 26.4 | S | 22.4 |
| Zn | 25.4 | C (diamond) | 6.1 |
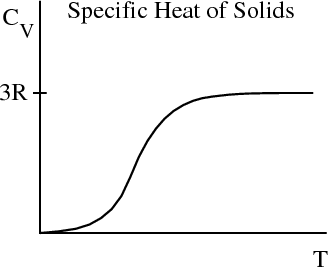
Note the obvious discrepancy with carbon (diamond). The classical theory predicts a temperature independent specific heat while all materials have the general property that cV is relatively temperature independent at high temperature but goes to zero as T → 0 as required by the third law of thermodynamics. Page 256 of Reif displays actual data for the specific heat of diamond. From the graph one can see that above about 1000 K, diamond has a specific heat of approximately 25 Joules/mole–K.
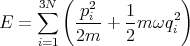 | (50) |
The partition function for N identical independent harmonic oscillators is
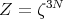 | (51) |
where
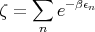 | (52) |
The energy levels of a single one dimensional harmonic oscillator are
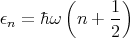 | (53) |
where n = 0, 1, 2,.... Then
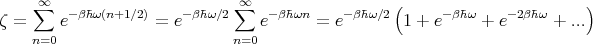 | (54) |
The sum is just an infinite geometric series where each term is obtained from the preceding one as a result of multiplication by e-βℏω. We can easily sum the series to get
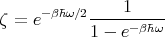 | (55) |
or
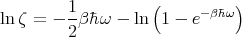 | (56) |
and
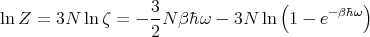 | (57) |
We can now obtain the mean energy:
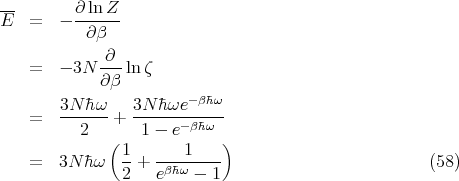
![--|
∂ E ||
CV = ∂T--||
--V|
∂-E-|| ∂-β
= ∂ β || ∂T
V ( --)
--1--- ∂-E-
= - kBT 2 ∂ β
[ Vβ¯hω ]
= - 3N-¯hω- - --e---¯h-ω--
kBT 2 (eβ¯hω - 1)2
( )2 β¯hω
¯h-ω-- ---e-------
= 3N kB kBT (eβ¯hω - 1 )2 (59)](lecture1164x.png)
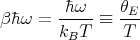 | (60) |
or
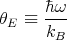 | (61) |
Then the molar specific heat becomes (with N = NA)
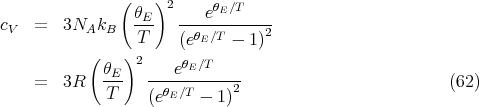
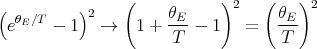 | (63) |
and
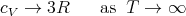 | (64) |
In the low temperature limit where T → 0 or θE∕T →∞
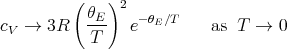 | (65) |
Careful experiments indicate that cV goes as T3 at low temperatures rather than exponentially. The explanation will come later when we consider the Debye theory of collective oscillations or normal modes of a crystalline solid consisting of a lattice of atoms.